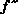 .
Here is a Harker section from such a map calculated using the anomalous
signal from a single Cu atom in the 96 residue metalloprotein CBP [Guss,
et al. 1989]:
.
Here is a Harker section from such a map calculated using the anomalous
signal from a single Cu atom in the 96 residue metalloprotein CBP [Guss,
et al. 1989]:
OK, you've collected your MAD data and run it through a phasing program that has produced initial estimates for the three quantities FT, FA, and Δφ. How do you get from there to a phase estimate for the protein, φT?
Since φT = φA + Δφ, if you had a value for φA then you'd be all set. To get this you need to locate the anomalous scattering atoms in the unit cell, so that you can calculate the phase φA of their contribution to the total scattering. The simplest way to do this is through Patterson analysis; it amounts to solving an n atom structure, where n is the number of anomalous scattering atoms.
There are many possible Patterson maps you might calculate using the data
you've collected. Of these, the most familiar may be the
Bijvoet Difference Patterson
, calculated with coefficients
ΔF2.
To increase the signal-to-noise ratio in your map you would logically choose
ΔF values from whichever of your
wavelengths λ has the largest expected
difference in F+ and F-, i.e.
the one with the largest .
Here is a Harker section from such a map calculated using the anomalous
signal from a single Cu atom in the 96 residue metalloprotein CBP [Guss,
et al. 1989]:
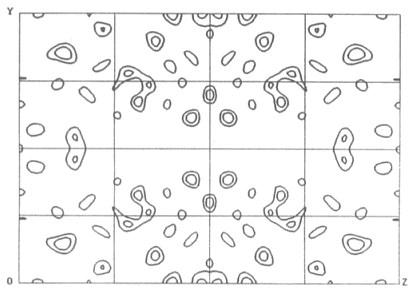 = 4.17e
= 4.17e
As you can see, it's rather noisy.
That's because even at the optimal wavelength the maximal value of
is only 4.2e.
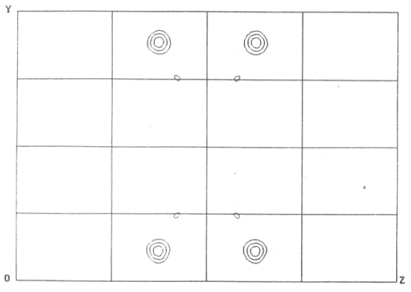
You can do much better than that, however. Rather than calculating any map
which uses only a subset of the data collected, you can instead use your
newly estimated value for |FA| to calculated a Patterson
map with coefficients
|FA|2.
Because all of the data you collected contributes simultaneously to this
map, it will be less noisy than a map calculated using only a subset of the
data:
If there are many anomalous scattering atoms in your structure then it may be difficult to interpret the resulting Patterson maps. 20 anomalous scattering atoms is about the upper limit.
The most successful approach developed to date for locating larger numbers of anomalous scattering atoms is the 'dual space' method implemented in the programs shake-n-bake (Smith et al, 1998) and shelxd (Sheldrick, 1998; shelx home page). These programs alternate cycles of work in real space (Patterson interpretation and placement of trial atomic positions) and work in reciprocal space (tangent refinement). Both programs have been used successfully to locate more than 50 Se sites in a SeMet substituted protein.
 back to index
back to index
 next topic
next topic